CanDLE: Illuminating Biases in Transcriptomic Pan-Cancer Diagnosis
Abstract
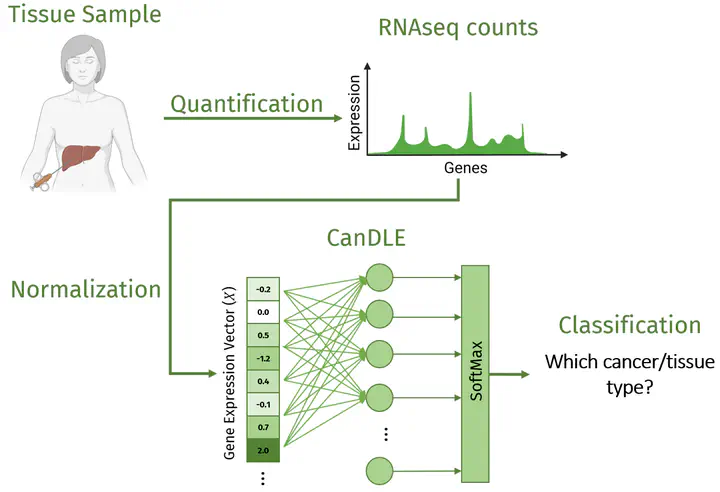
Automatic cancer diagnosis based on RNA-Seq profiles is at the intersection of transcriptome analysis and machine learning. Methods developed for this task could be a valuable support in clinical practice and provide insights into the cancer causal mechanisms. To correctly approach this problem, the largest existing resource (The Cancer Genome Atlas) must be complemented with healthy tissue samples from the Genotype-Tissue Expression project. In this work, we empirically prove that previous approaches to joining these databases suffer from translation biases and correct them using batch z-score normalization. Moreover, we propose CanDLE, a multinomial logistic regression model that achieves state of the art performance in multilabel cancer/healthy tissue type classification (94.1% balanced accuracy) and all-vs-one cancer type detection (78.0% average max F1).